跟随性药物:前人栽树后人乘凉 一代新药胜旧药
首创药物是通过寻找全新的药物靶标、作用机制和分子结构,以便达到攻克无药可医的重大疾病难题的目的,从靶标的发现、苗头分子的确定、苗头分子向先导物的过渡、先导化合物的确定与优化、成药性优化到靶标再验证往往需要数十年的研发时间,需要十年磨一剑的攻坚精神。跟随性药物则是“前人种树、后人乘凉”,一定程度上有规可循,却也有自身的特点,所以市场上也出现很多优于首创药物的后继药物。
提高半衰期和长效性
1986年FDA批准首创毒蕈碱乙酰胆碱受体3(M3受体)拮抗剂异丙托溴铵上市,用于治疗慢性阻塞性肺病,常用剂量为每日吸入3~4次,每次20~40μg,半衰期为1.6~3.6h。引入噻吩结构,增强其疏水性,得到后继药物噻托溴铵,常用剂量为每日吸入1次,每次18μg,半衰期为5~6d。
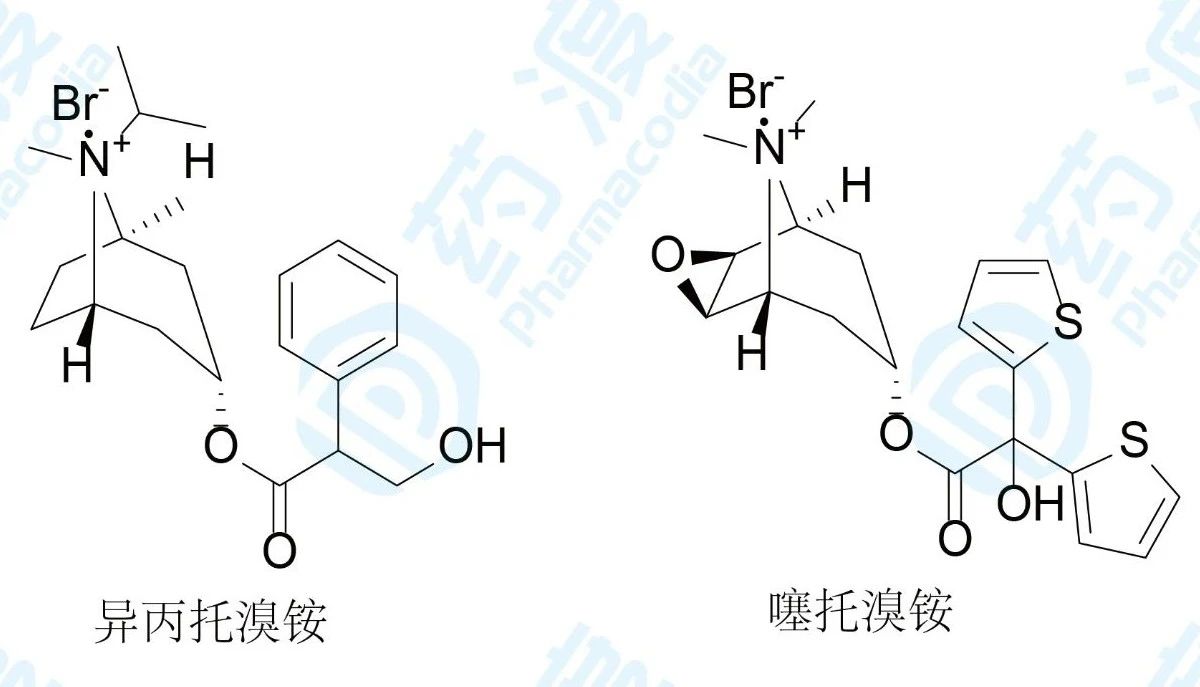
表1 异丙托溴铵和噻托溴铵的结合动力学性质及选择性
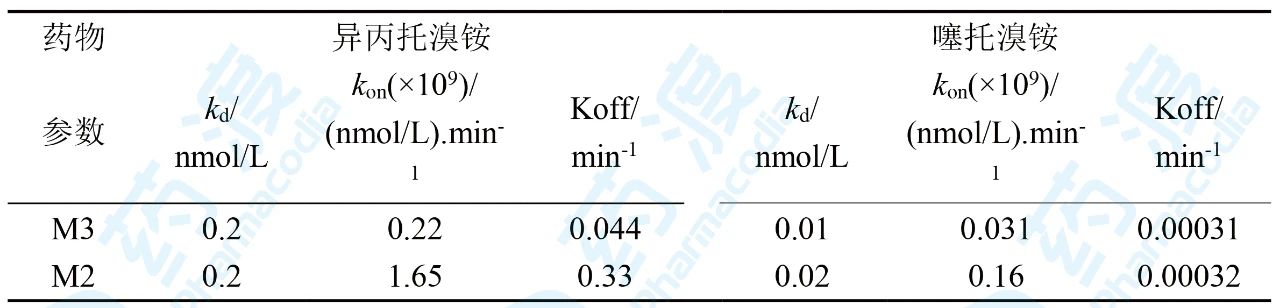
对比二者的生物参数发现,噻托溴铵与M受体的结合力(kd)强于异丙托溴铵,且对M3选择性提高,另外噻托溴铵对M3受体的离解速率(koff)显著的低于异丙托溴铵的离解速率,从而噻托溴铵在M3受体上的驻留时间显著延长,停留并结合在M3的时间长,从而用药剂量和频度少于异丙托溴铵。
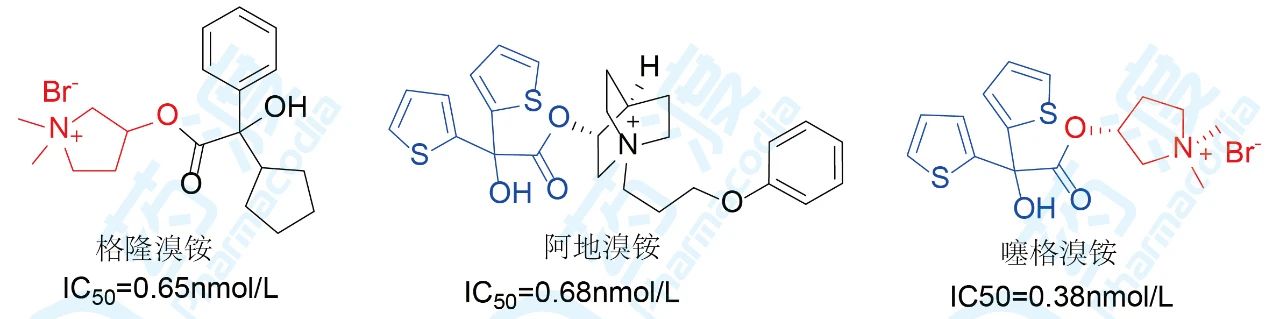
格隆溴铵和阿地溴铵对M受体的选择性差,虽然格隆溴铵的血浆稳定性较好,但是容易产生口感、便秘等副作用,严重甚至导致心脏毒性,而阿地溴铵则血浆稳定性差,需要大剂量给药。孙宏斌研究团队结合噻托溴铵和阿地溴铵的优势骨架,并进行系统的构效关系研究得到对M3受体拮抗作用更强的噻格溴铵。
改变用药途径和适应症
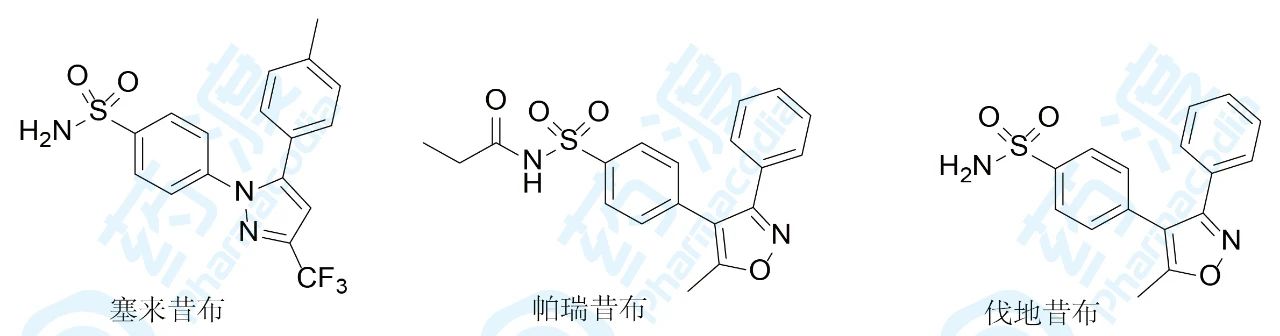
1999年首创抗炎药塞来昔布的上市,使得辉瑞收获了极大的财富。而辉瑞的第二代特异性COX-2抑制剂帕瑞昔布作为一种以钠盐形式注射的COX-2抑制剂更是让患者在术后急性痛的方面拥有了更多和更好的选择。帕瑞昔布在体内经代谢水解生成伐地昔布起效,所以是前药。分子中的磺酰氨基经丙酰化,氨基呈酸性(双酰基拉电子),可制成可溶性钠盐。释放出的伐地昔布与塞来昔布有相同的药效团特征,只是咪唑环骨架变为异噁唑环。作为唯一注射用的COX-2抑制剂,帕瑞昔布改变了适应症,用于解除围术期中度或重度疼痛。
增强药理强度
GSK研发的安普那韦于1999年批准上市,用于治疗艾滋病,7年后Tibotec公司在安普那韦四氢呋喃环上再并合一个环成为双环,得到的地瑞那韦活性提高近100倍。分子对接发现,新并合环的氧原子可与天冬氨酸残基形成两个氢键,形成的氢键网络增高了与靶标的结合力。热力学分析发现,氢键提供的结合能(ΔG)主要是焓贡献(ΔH),由于焓比熵(−TΔS)更显示结合的特异性,地瑞那韦结合强度和特异性更强。表2数据表明,安普那韦的焓熵贡献各占一半,而地瑞那韦则主要是焓贡献。
表2 安普那韦和地瑞那韦的活性及热力学分析

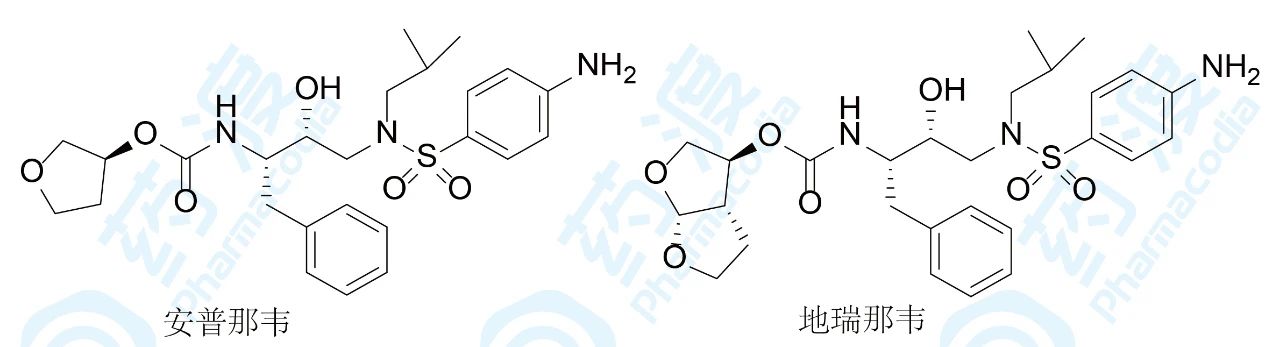
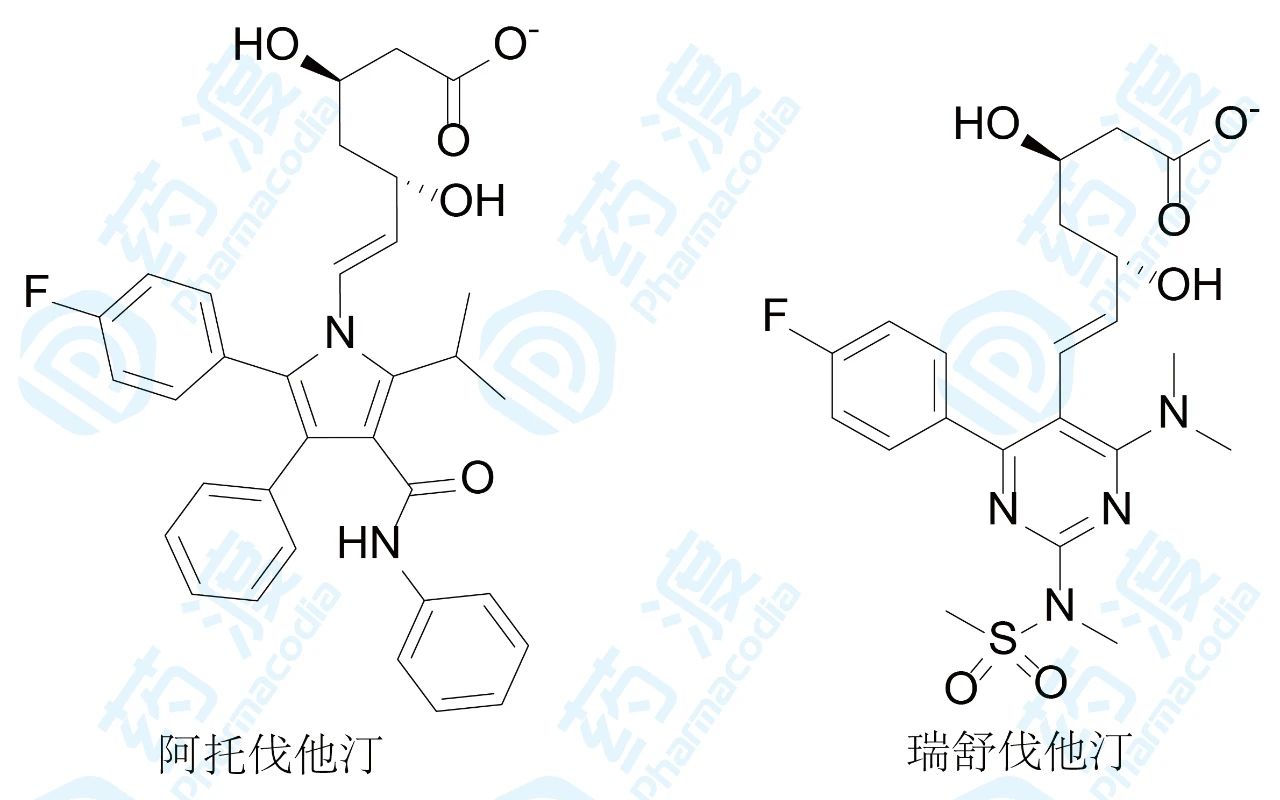
增加氢键结合和范德华作用比疏水性结合具有更强的特异性,据此,跟随性药物可提高药理作用的特异性,也避免代谢的复杂性。阿托伐他汀和瑞舒伐他汀与HMGCoA还原酶的结合是这方面的实例。阿托伐他汀和瑞舒伐他汀是第5和第6个上市的“他汀”类药物,临床应用和市场份额比4个前驱药物优胜,重要原因是结构因素具有特点。在阿托伐他汀和瑞舒伐他汀的分子的下半部的疏水片段上分别引入了酰胺基和磺酰基片段,增加了氢键的结合,该特异性结合反映在阿托伐他汀和瑞舒伐他汀与酶结合能的组成上,焓贡献是主要驱动力,而前4个他汀则是以熵驱动为主的结合。
改善药动学
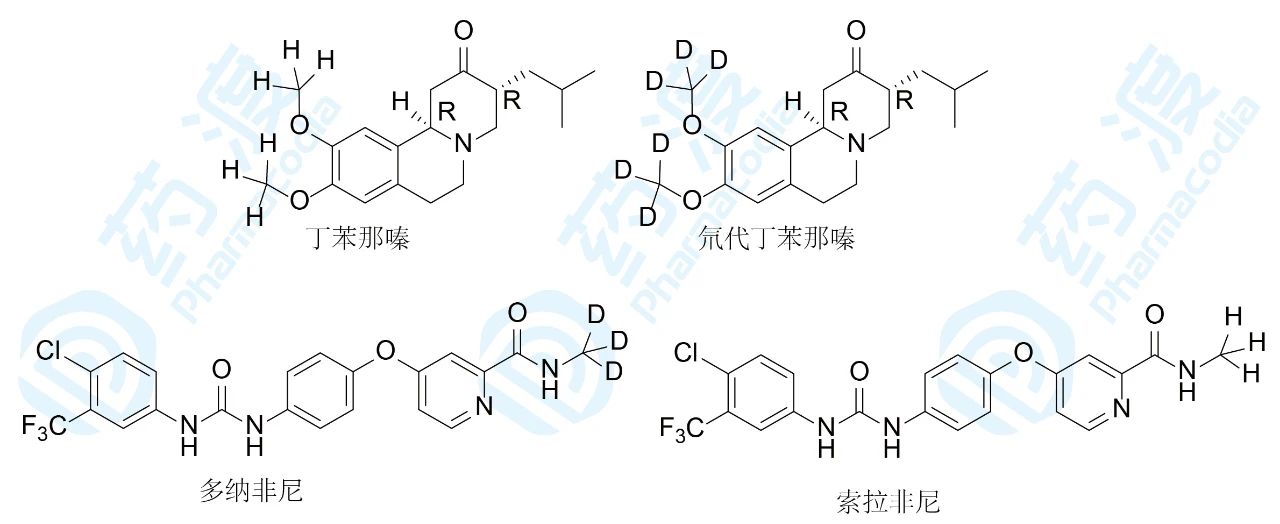
丁苯那嗪是20世纪50年代已知的化合物,直到2008年FDA批准用于治疗亨廷顿氏病。其作用机制是靶向抑制囊泡单胺转运蛋白2,阻断并抑制单胺类神经递质的储存,减少单胺对突触小泡的摄取。口服丁苯那嗪在肝脏被CYP2D6氧化脱甲基,首过效应导致生物利用度低。代谢环节是甲氧基的C-H被氧化,由于C-D键强于C-H。Auspex公司根据动力学同位素效应(kH/kD=6.5),研发了全球首个氘代药物——氘代丁苯那嗪,2017年FDA批准用于治疗中枢神经性疾病亨廷顿氏病等舞蹈病。氘代丁苯那嗪只在代谢位点作氘代,并用光学活性的R,R-异构体,提高了代谢稳定性,降低了用药剂量和频次。
我国的泽璟生物公司研发的多纳非尼目前处于Ⅲ期临床阶段,是拜耳首创的索拉非尼的跟随性氘代物。多纳非尼治疗晚期肝细胞癌患者的Ⅱ/Ⅲ期试验显示,多纳非尼0.2g/次组和0.3g/次组的中位总体生存时间分别长达12.6个月和11.8个月。其中在第16周,多纳非尼0.2g/次组(Ⅲ期临床试验剂量)和0.3g/次组经独立影像学评估的疾病控制率分别为42.5%和40.9%,疗效差异无统计学意义。安全性方面,多纳非尼0.2g/次组和0.3g/次组维持初始剂量的中位生存时间分别为90d和72d,总体安全性良好。此研究表明,多纳非尼0.2g/次的口服剂量或将成为晚期肝细胞癌治疗的新一线用药。
克服耐药性
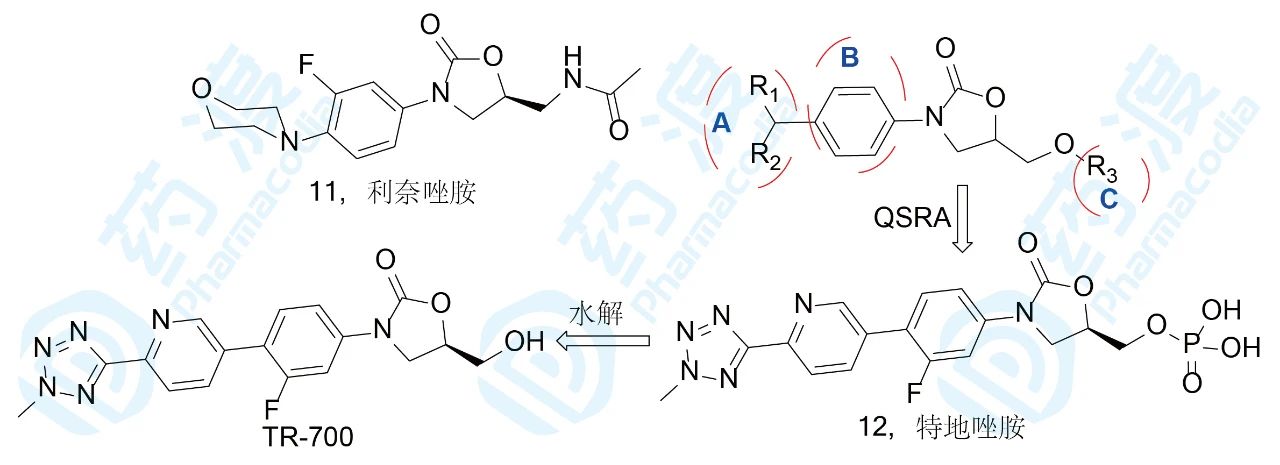
利奈唑胺是细菌蛋白合成的抑制剂,作用环节是在核糖体上阻断mRNA翻译成蛋白的初始阶段,结合部位是核糖体50S亚基的A位点。由于抑菌活性强而广谱,吸收好(口服生物利用度F=100%),鲜有跟随性药物上市,直到2014年第二个跟随性药物特地唑胺成功。特地唑胺是以磷酸酯为前药,可口服或注射用药,在血浆中被磷酸酶水解掉磷酸基成活性形式(TR-700)。对耐受甲氧西林的革兰阳性菌的抑制作用强于利奈唑胺4~16倍。特地唑胺的研制路径是在分析大量的构效关系的基础上成功的:A部分上引入氮杂环后副作用降低;B部分引入F原子则抗菌活性提高,其他取代基活性降低;C部分的大位阻活性降低。
跟随性药物的研发是对首创药物的补充和深入,有首创药物的影子,但也具有自身独特性。随着我国综合国力的不断提高,基础研究水平的日益完善,希望越来越多的药物尤其是首创药物和具有独特优势的跟随性药物成为“中国制造”。
参考资料
1.Copeland RA. Conformational adaptation in drug-target interactions and residence time [J]. Future Med Chem, 2011, 3: 1491−1501.
2.Heretsch P, Tzagkaroulaki L, Giannis A. Modulators of the Hedgehog signaling pathway [J]. Bioorg Med Chem, 2010, 18: 6613-6624.
3. Taipale J, Chen JK, Cooper MK, et al. Effects of oncogenic mutations in smoothened and patched can be reversed by cyclopamine[J]. Nature, 2000, 406:1005-1009.
4. Talley JJ, Bertenshaw SR, Brown DL, et al. N-[[(5-Methyl-3-phenylisoxazol-4-yl)-phenyl] sulfonyl]propanamide, sodium salt, parecoxib sodium: a potent and selective inhibitor of COX-2 for parenteral administration [J]. J Med Chem, 2000, 43: 1661−1663.
5. Tie YPI, Boross YF, Wang L, et al. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multidrug-resistant clinical strains[J]. J Mol Biol, 2004, 338: 341−352.
6. Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (lapatinib). Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells[J].Cancer Res, 2004, 64: 6652−6659.
7. Citrome L. Breakthrough drugs for the interface between psychiatry and neurology [J]. Int J Clin Pract, 2016, 70: 298-299.
8. Locke JB1, Finn J, Hilgers M, et al. Structure-activity relationships of diverse oxazolidinones for linezolid-resistant Staphylococcus aureus strains possessing the cfr methyltransferase gene or ribosomal mutations [J]. Antimicrob Agents Chemother,2010, 54: 5337−5343.

- 聚力拓市启新程 吉享绿城焕生机
— 王老吉药业亮相广药南宁招商大会 以 “签约赋能 + 嗦粉...
- 国内首个治疗良性前列腺增生复方制剂“爱廷
3月22日下午,爱廷列“基层前列腺健康行”公益项目启动会暨上...
- 仑卡奈单抗和多奈单抗的巅峰对决!
目前医学界普遍认为,大脑中β淀粉样蛋白(Aβ)的异常沉积是阿...
- 仑卡奈单抗上市已一年,中国阿尔茨海默病治
在全球老龄化加速的背景下,阿尔茨海默病(Alzheimer’...
