重磅!我国科学家揭示CAR-T细胞触发细胞因子释放综合征机制
尽管用经过基因改造后表达嵌合抗原受体(CAR)的T细胞(CAR-T细胞)治疗B细胞恶性肿瘤的临床应用取得了成功,但是细胞因子释放综合征(CRS)却阻碍了这种疗法在患者中的有效性。众所周知,CRS是由急性炎症反应触发的,其特征是发烧、低血压和与血清细胞因子升高有关的呼吸功能不全。尽管人们已报道在经过CAR-T细胞治疗的人源化小鼠模型中,巨噬细胞参与了CRS的发病机制,但触发CRS的机制尚不清楚。输注到患者体内的CAR-T细胞经历活化和增殖,快速增殖的CAR-T细胞可能会在短时间内导致B白血病细胞迅速大量死亡。巧合的是,急性淋巴细胞白血病(ALL)患者的疾病负担也与CRS的发生率和严重程度密切相关。如此大规模的恶性B细胞死亡参与CRS发病机制的方式仍不明确。
细胞可以经历不同类型的死亡。细胞凋亡最初被认为是唯一的受到调控的程序性死亡形式。但是,近期研究已证实了一种以前未被识别的程序性坏死,其特征是细胞快速肿胀、质膜上出现较大的气泡以及促炎因子释放。至少已鉴定出两种程序性坏死细胞死亡途径,包括MLKL介导的坏死性凋亡(necroptosis)和GSDMD(Gasdermin D)或GSDME(Gasdermin E)介导的细胞焦亡(pyroptosis)。
将RIPK1招募到TNF-α受体上可与RIP3形成一种死亡复合物,随后产生膜纳米孔,从而导致坏死性凋亡。不同于MLKL的是,GSDMD或GSDME由炎性半胱天冬酶(caspase 1、caspase 4、caspase 5和小鼠caspase 11)或caspase 3激活,可形成插入到细胞膜中的寡聚物而形成纳米孔,因而介导细胞焦亡。
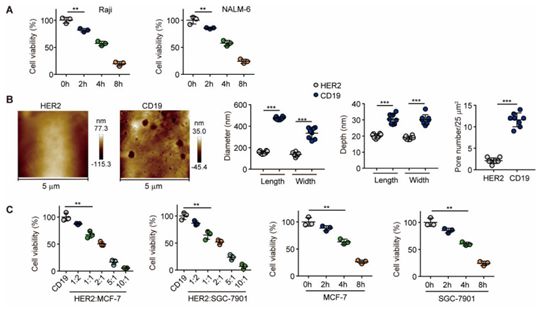
在一项新的研究中,来自中国医学科学院、郑州大学、华中科技大学和北京大学的研究人员提供的证据表明,人类B白血病细胞和其他靶肿瘤细胞表达了足够数量的GSDME,所表达的GSDME可被由CAR-T细胞释放的颗粒酶B激活的caspase 3高效激活,从而导致靶细胞遭受细胞焦亡。细胞焦亡释放的因子刺激巨噬细胞产生促炎性细胞因子,这很可能触发接受CAR-T细胞治疗的患者中发生的CRS。相关研究结果发表在2020年1月17日的Science Immunology期刊上,论文标题为“Gasdermin E–mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome”。
图片来自Science Immunology, 2020, doi:10.1126/sciimmunol.aax7969
这些研究人员发现CAR-T细胞由于释放大量穿孔素和颗粒酶B而激活了B白血病细胞中的caspase 3-GSDME途径,从而导致细胞焦亡和随后的CRS。已观察到CAR-T细胞经历增殖过程并在体内的某个时间点达到极高的增殖频率。因此,在相对较短的时间内,大多数靶细胞可能会经历细胞焦亡,导致受到激活的巨噬细胞通过活化的caspase 1产生IL-6和IL-1β,从而触发CRS。阐明这种分子机制为CRS严重程度与CAR-T细胞在治疗期间的数量和B白血病细胞负荷有关的临床观察提供了深刻的见解。
在人类B白血病细胞、MCF-7乳腺癌细胞和小鼠B16黑色素瘤细胞中检测到GSDME表达是出乎意料的,这是因为它作为一种孔形成蛋白发挥作用,而且它的激活是潜在危险的,并且可能导致细胞死亡。据报道,GSDME在许多检测到的肿瘤细胞系中不表达。与此相一致的是,GSDME基因的启动子区域显示出高甲基化状态,这就表明GSDME基因在细胞中遭受表观遗传沉默。 GSDME被认为是一种通过caspase 3裂解诱导程序性细胞死亡的肿瘤抑制基因,并且肿瘤细胞可能已进化出表观遗传学手段来沉默GSDME表达,从而允许肿瘤发生。然而,GSDME在B白血病细胞和其他肿瘤细胞中的高表达提示着GSDME很可能对肿瘤细胞中的孔形成发挥替代作用。人们目前正在研究GSDME表达克服高甲基化调控的方式以及GSDME是否具有除孔形成以外的常规功能目前正在处于研究当中。
这项研究的重要发现是CAR-T细胞比未转导的天然T细胞释放更多的穿孔素/粒酶B。T细胞释放溶细胞性效应分子取决于两种信号的激活:MHC-抗原肽-TCR(信号1)和CD80/CD86-CD28(信号2)。信号2的完全激活取决于TCR信号的激活强度。TCR信号激活的轻链激酶(LCK)使得CD28酪氨酸残基发生磷酸化;与此同时,TCR信号激活的LAT和SLP-76磷酸化和激活关键的CD28下游信号分子PLC-γ,因而将磷脂酰肌醇4,5-双磷酸(PIP2)降解为二酰甘油(DAG)和1,4,5-三磷酸肌醇(IP3)。
基于对T细胞激活的了解和基因工程取得的进步,这些研究人员设计出针对人类T细胞的合成CAR。设计CAR的基本概念是将单链可变区片段(scFv)与CD3ζ细胞内信号转导模块连接在一起,以在抗原结合后诱导T细胞激活。当前,这种模块化结构已从单个CD3ζ信号结构域扩展到CD3ζ-CD28、CD3ζ–4-1BB或CD3ζ–CD28–4-1BB信号结构域,以模拟信号1和信号2。
鉴于CAR与其抗原之间的亲和力可能比TCR与MHC-肽复合物之间的亲和力高100倍,因此这种优越的亲和力加上共刺激信号使得CAR-T细胞能够释放大量的穿孔素/粒酶B,这是CAR-T细胞介导的靶细胞经历细胞焦亡所必需的。一旦进入细胞质后,颗粒酶B可以将caspase 3的无活性前体(procaspase 3)裂解为它的活性形式。活化的caspase 3诱导细胞凋亡或裂解GSDME,这种理解通过膜孔形成触发细胞焦亡。但是,细胞具有快速修复质膜中形成的孔的能力。GSDME是否触发细胞焦亡取决于膜孔形成与膜修复之间的平衡。尽管具有相同的GSDME数量,但是天然的TCR CD8+ T 细胞仅导致低水平的GSDME裂解,而CAR-T细胞释放出更高水平的穿孔素/粒酶B,因而导致更多的GSDME激活。
由于释放富含损伤相关分子模式(DAMP)分子的细胞质内容物,细胞焦亡中发生的裂解具有高度促炎性。在这项研究中,这些研究人员证实肿瘤细胞经历的细胞焦亡导致巨噬细胞中的caspase 1和GSDMD的激活,从而导致大量促炎细胞因子的释放和CRS的发生。在这些促炎细胞因子中,IL-6和IL-1β尤为重要。临床上,中和IL-6的抗体被广泛用于预防和/或治疗接受CAR-T细胞治疗的患者中发生的CRS。作为多效细胞因子,IL-6主要受到NF-κB、AP-1和STAT3等转录因子的调节。诸如脂多糖(LPS)之类的病原体相关分子模式(PAMP)分子和诸如热休克蛋白(HSP)和HMGB1之类的DAMP分子可通过激活这些转录因子来刺激巨噬细胞产生IL-6。
这些研究人员发现HMGB1存在于细胞焦亡的上清液中,因而直接激活巨噬细胞中的IL-6。 IL-1β是以前体形式合成的,它的释放取决于caspase 1的激活,caspase 1是一种受到炎性体严格调节的促炎性半胱天冬酶。炎性体NLRP3已被包括微生物毒素、颗粒物、晶体、β-淀粉样蛋白聚集物或细胞外ATP在内的多种刺激物广泛激活。
在这项研究中,这些研究人员发现细胞焦亡的上清液含有ATP,用P2X7受体拮抗剂处理或降解ATP可抑制细胞焦亡的上清液激活巨噬细胞中caspase 1的能力。在他们开发的小鼠CRS模型的帮助下,他们进一步证实不论是敲除靶肿瘤细胞中的GSDME,也不论是剔除巨噬细胞,还是抑制caspase 1/GSDMD都可阻止CRS的发生。阐明这种分子途径对于更好地了解与CAR-T细胞疗法相关的毒性至关重要。2018年,Verena Staedtke等人已发现儿茶酚胺阻断剂可以抑制巨噬细胞释放促炎性细胞因子。因此,对GSDME和儿茶酚胺的联合阻断可以在不降低肿瘤清除的情况下更好地治疗CRS。
尽管这项研究报道CAR-T细胞通过GSDME依赖性途径诱导靶肿瘤细胞遭受细胞焦亡,但也可能存在其他途径来介导CAR-T细胞引起的靶细胞焦亡。近期研究已报道CAR-T细胞可能动员TNF-α来介导这种杀伤过程,不过这可能与颗粒酶B和穿孔素无关。这项发现的一种可能性是CAR-T细胞可以使用一种两步骤策略来攻击靶细胞。颗粒酶B和穿孔素引发了第一波杀伤,如果靶细胞逃脱了第一波攻击,那么TNF-α就会发起第二波杀伤。这也可能解释了为何细胞焦亡可在不显著影响CAR-T细胞介导的杀伤作用的情况下不被阻断。当前的这项研究揭示了CAR-T细胞和天然的TCR-T细胞导致的细胞死亡类型之间存在机制上的差异,这就为通过将肿瘤细胞死亡从细胞焦亡切换到细胞凋亡来修饰CAR以减少CRS提供了机会。
参考资料:
Yuying Liu et al. Gasdermin E–mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Science Immunology, 2020, doi:10.1126/sciimmunol.aax7969.


- 礼进生物将在2023 ASCO上报告4-
礼进生物将在2023年美国临床肿瘤学会 (ASCO) 年会上...
- 脑静脉血栓实现智能化诊断,脑卒中AI研究
近日,推想医疗与首都医科大学附属北京朝阳医院放射科杨旗教授团...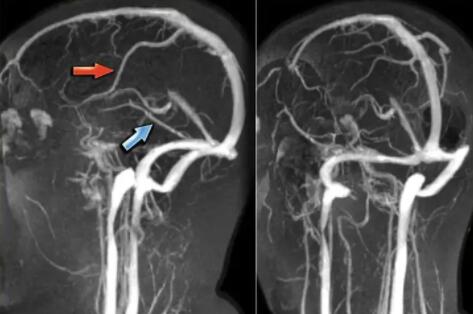
- 科兴制药产业布局再发力 探索合成生物技术
11月2日,科兴制药(股票代码:688136)发布公告,公司...
- 拜耳转移乳腺癌ErSO疗法尚未进入临床试
此前有报道称拜耳一种小分子药物ErSO对动物体内的癌细胞有9...